Controls, controls, controls!
Over the last two decades, extensive technological and methodological advances have led to the widespread use of mass spectrometry (MS)-based bottom-up proteomics, which involves the digestion of proteins into peptides followed by identification and quantification of the resulting peptides by LC-MS. MS based analysis is the method of choice for large-scale exploration of proteomes in biological systems and has significantly contributed to the comprehension of a variety of biological processes. In the field of clinical research, quantitative MS has been an integral part of numerous biomarker discovery and evaluation studies1.
A typical bottom-up proteomics mass spectrometry experiment consists of multiple different phases including sample preparation, liquid chromatography, mass spectrometry, and bioinformatics. Mass spectrometry-based proteomics experiments can be subject to variability2,3 within each of the 4 main stages impacting the results. The scheme below is representing variability in bottom-up mass spectrometry.

Fig 1. Variability in proteomics analyses can come from a lot of places. LC, sample preparation, bioinformatics and the MS analysis itself can contribute to variability in the results. Inspired by Bittremieux W et al 2018 (2).
Ideally, to achieve high-quality and highly reproducible results, all steps in this procedure must be carefully validated and controlled with appropriate quality control reagents and protocols. Whilst there are many procedures that are routinely used to standardize LC-MS hardware performance and bioinformatic data analysis, less emphasis seems to be placed on ensuring that all aspects of sample preparation are correctly controlled for. This can lead to the classic scenario of “put rubbish in, get rubbish out” i.e; expensive, high performance LC-MS system has a sample of sorts injected and then the data analysis software is expected to sift it for the valuable snippets of information which cannot exist. Put simply, robust sample preparation protocols underpinned with quality control reagents to directly standardize specific steps will clearly increase the overall consistency and robustness of the data, reduce the overall experimental time and save precious LC-MS instrument time. Furthermore, standardization of the overall proteomics workflow increases the ease of exchange of reproducible data and protocols between scientific groups.

For robust sample preparation methods in proteomics experiments that are used to identify and quantify thousands of proteins within complex matrices, such as tissue or plasma, certain common steps are performed. These steps are protein extraction followed by reduction, alkylation, clean-up step and enzymatic digestion.
Bottom-up proteomic experiments routinely use enzymatic digestion of large numbers of proteins derived from complex matrices to produce proteotypic peptides for LC-MS analysis. The enzymatic proteolysis step is of paramount importance as only efficient digestion of the multiple proteins will ensure production of the correct proteotypic peptides in stoichiometric amounts for accurate quantification. The proteolytic digestion can be performed on thousands of different proteins that are present in different abundancies and crucially, each protein behaves differently to enzymatic digestion; some will be easy to digest; some difficult and many will be in that intermediate area. Incomplete digestion will thus impair the qualitative and quantitative results of such experiments and it is difficult to recognize this experimentally, unless there is a reagent that can clearly show enzymatic digestion reflecting those that can be digested with ease, difficulty or somewhere in between. The situation is further complicated because there are numerous enzymatic digestion protocols within the literature and, anecdotally, even different ones within the same research group. Added to this is there are many commercially available sources of trypsin with nominally stated “grades”, variable specifications and different experimental protocols based on protein samples that are often not representative to the plethora used in the multitude of proteomic labs4. Using a dedicated reagent that has been designed to specifically monitor and standardize digestion efficiency can offer clear value by increasing workflow reproducibility, gaining critical data quality and saving costly LC-MS and sample preparation time derived from inadequately digested sample analysis
We have listed some existing solutions below for proteomics quality control with their main characteristics:
1) DIGESTIF from Promise Proteomics:
Description:
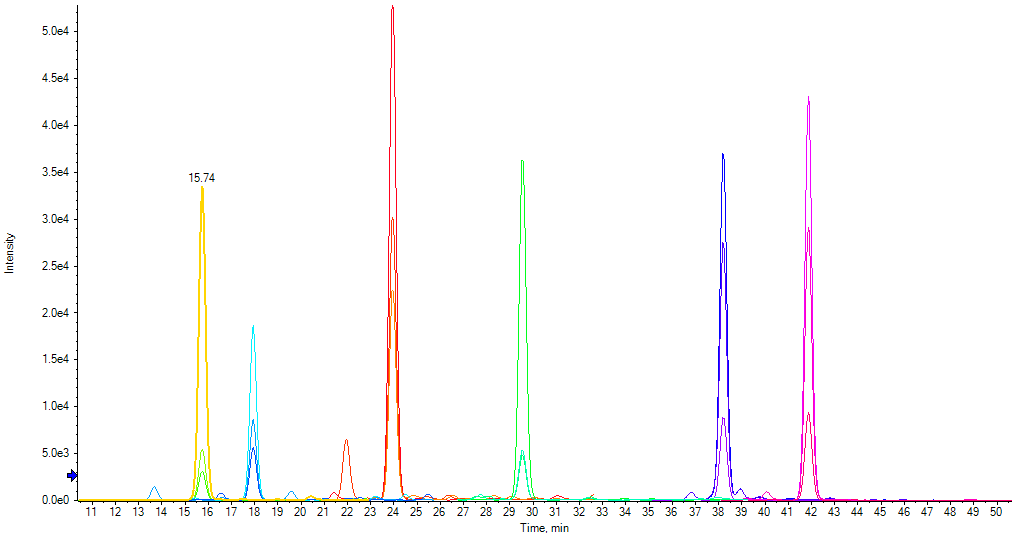
DIGESTIF is a synthetic Stable Isotope Labelled protein, expressed in E. coli. The DIGESTIF standard consists of a soluble recombinant protein scaffold to which a set of 11 artificial peptides (iRT peptides) with good ionization properties has been incorporated. In the protein scaffold, the amino acids flanking iRT peptide cleavage sites were selected to either favor or hinder protease cleavage. In this way, incorporation of 11 iRT peptides means that Digestif has been specifically designed as a model reflecting the digestion properties of a broad range of proteins that will occur in the proteome of a complex sample. Digestif is thus analyzed by LC-MS and benefits from the readout’s increased specificity showing a peak for each released peptide
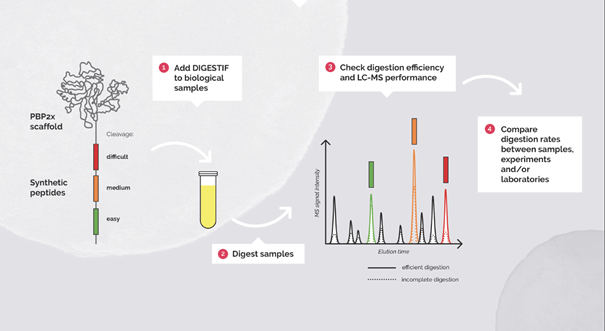
Fig 2. Workflow for the usage of Digestif as a digestion and retention time control.
+ : An added benefit is that the retention time and intensity of iRT peptides released during digestion can be used to check LC-MS performance saving further time and cost if there are hardware issues.
- : Require exploratory experiments to define optimized condition of use.
Description: QCAL5 is a protein, a concatenation of tryptic peptides that is expressed in E. coli. QCAL is a MS calibration standard that is built from 22 peptides, ranging from ~ 410- 3100 Da and is designed to provide standards for peptide separation by reversed-phase chromatography.
+ : After trypsin digestion, QCAL provides a stoichiometrically controlled peptide mixture allowing the concomitant assessment and optimization of multiple MS-instrument parameters on a wide variety of instrument platforms1.
- : Such synthetic cleavage sites are by their very nature, in vitro, in function. Simply; the peptides released from the QCAL are not in the same sequence context as peptides released from the endogenous proteins. Complex protein mixtures not only consist of proteins that are easily cleaved but also contain highly structured species that may, in part, resist efficient proteolysis. QCAL is not predicted to form significant regions of secondary structure. In fact, they are designed and optimized for the most efficient release of their surrogate peptides. While this may be appropriate for certain specifically targeted proteins, it means that they are poorly suited for evaluation of the digestion efficiency of the plurality of proteins contained within complex mixtures1.
3) JPT Peptide TrypCheck Kit Fluorescence.
Description: It is a fluorescent peptide kit consisting of 3 Förster Resonance Energy Transfert (FRET) peptides with trypsin cleavage sites, 1 fluorescent control peptide for quantification.
A peptide bearing one or more Arg or Lys in the center of its sequence covalently connects a Abz fluorophore and its respective Nitrotyrosine quencher. Upon tryptic digestion, the quencher is cleaved from the peptide, releasing the fluorophore which can undergo stokes shift upon excitation to produce the emitted light that can be analysed. The amount of the released fluorophore has a correlation with the cleavage efficiency of the digestion. This product requires fluorescence-reading instrumentation.
+ : Fluorescence instrumentation is fast and easy to use.
- : Whilst the use of a fluorescence read should make this sensitive to quantify low concentrations of peptides produced from the digestion, it is an indirect labelling measurement of tryptic digestion and subject to the challenges that occur with all fluorescence assays; such as high background caused by matrix, photobleaching; temperature or pH stability and assay interferences. The fluorescence readout does not have the specificity of the LC-MS readout and since the JPT peptides cannot be used in the injected sample, they cannot be used to check for retention time and LC-MS performance.
Since there are only 3 peptides; again, there are too few digestion events to reflect the proteomic sample complexity of protein number; digestion efficiency and abundance.
4) Pierce™ Digestion Indicator for Mass Spectrometry
Description: The Thermo Scientific Pierce Digestion Indicator for Mass Spectrometry is a non-mammalian recombinant protein (26kDa) with five signature peptides for use in determining the digestion efficiency and reproducibility across multiple samples. This product is sufficient for production of five signature peptides upon digestion for mass spectrometry.
The Pierce Digestion Indicator serves as an internal digestion control standard protein to ensure protocol performance and to quantify sample preparation processing and digestion efficiency across samples.
+ : Non-mammalian – can be easily distinguished from endogenous mammalian peptides
- : Having only 5 peptides may not be sufficient for a complete monitoring over the full chromatogram.
5) Universal Proteomics Standards (UPS)
Description: The Universal Proteomics Standard (UPS1) and the Proteomics Dynamic Range Standard (UPS2) products are complex, well-defined, well characterized reference standards for mass spectrometry. Both standards contain the same 48 human proteins, ranging in molecular mass from 6,000 to 83,000 Daltons.
+ : Good standard for monitoring dynamic range of the LC-MS performance for proteomics
- : Some of the proteins in the mix can be endogenous and/or contaminating. The resulting peptides could thus interfere with the quantification of some proteins.
Description: The full-length BSA precursor protein is 607 amino acids in length. An N-terminal 18-residue signal peptide is cut off from the precursor protein upon secretion, hence the initial protein product contains 589 amino acid residues. An additional six amino acids are cleaved to yield the mature BSA protein that contains 583 amino acids.
+ : BSA is a stable, moderately non-reactive protein.
- : Bovine serum Albumin is routinely used in laboratories for a variety of applications, such as a Western blot blocking protein or as a major additive in tissue culture media used to grow cells that are subjected to proteomic experiments. This has led it being considered a contaminant of proteomic workflows in the same way that Keratins are: its use as a QC depends on the sample being completely free of BSA so complete digestion of a specific concentration can be assessed. BSA is also a single protein which does not produce proteotypic peptides that span the entire proteomic chromatogram that occurs when thousands of proteins are digested and analyzed. So, using a possible contaminant as a QC means it is difficult to correctly use as an optimal QC step.
Concluding remarks
There you have it. As in any scientific experiments, mass spectrometery proteomics workflow need to include several control steps to ensure maximal reproducibility. Correctly monitoring the digestion step, as well as the chromatography, can help to tremendously increase the overall quality of one's proteomics experiment. Many great products are available to do so, and we strongly believe in their efficiency.
**Disclosure**
This post was written prior to the agreement with Promise Advanced Proteomics for distribution of their Digestif products.
Refs :
1. Dorothée Lebert, Mathilde Louwagie, Sandra Goetze, Guillaume Picard, Reto Ossola, Caroline Duquesne, Konrad Basler, Myriam Ferro, Oliver Rinner, Ruedi Aebersold, Jérôme Garin, Nicolas Mouz, Erich Brunner, and Virginie Brun. DIGESTIF: A Universal Quality Standard for the Control of Bottom-Up Proteomics Experiments.J. Proteome Res. 2015, 14, 787−8032. Bittremieux W, Tabb DL, Impens F, Staes A, Timmerman E, Martens L, Laukens K. Quality control in mass spectrometry-based proteomics. Mass Spectrom Rev. 2018 Sep; 37(5):697-711.
3. Glatter, T., Ludwig, C., Ahrné, E., Aebersold, R., Heck, A. J. R., Schmidt, A. Large-Scale Quantitative Assessment of Different In-Solution Protein Digestion Protocols Reveals Superior Cleavage Efficiency of Tandem Lys-C/Trypsin Proteolysis over Trypsin Digestion. J. Proteome Res. 2012, 11, 5145-5156
4. Burkhart, J. M., Schumbrutzki, C., Wortelkamp, S., Sickmann, A., Zahedi, R. P., Systematic and quantitative comparison of digest efficiency and specificity reveals the impact of trypsin quality on MS-based proteomics J. Proteomics 2012, 75(4), 1454-1462.
5. Eyers CE, Simpson DM, Wong SC, Beynon RJ, Gaskell SJ. QCAL--a novel standard for assessing instrument conditions for proteome analysis. J Am Soc Mass Spectrom.2008 Sep;19(9):1275-80.