Votre panier est actuellement vide !
Détails de la ressource
Comment fonctionne l’identification des protéines par LC-MS/MS ?
par Jean-Philippe Couture, PhD, Gestionnaire des ventes scientifiques
Comment identifier une protéine inconnue avec la spectrométrie de masse ?
Nous sommes tous passés par là. Les recherches vont bon train, les résultats arrivent et une image de la façon dont tout fonctionne ensemble commence à se dessiner. Puis, on se heurte à un mur : une protéine inconnue gênante au milieu du modèle.
Le projet s’est déroulé sans encombre, à l’exception de ce point d’interrogation. Et maintenant, votre chercheur principal vous demande d’identifier l’acteur manquant pour résoudre le mystère. Vous allez donc sur Google ou PubMed et commencez à chercher des moyens d’identifier des protéines inconnues. La réponse à votre question est claire : vous avez besoin de la spectrométrie de masse.
Mais vous ne savez pas comment il fonctionne et vous devrez l’expliquer lors de la prochaine réunion du laboratoire.
Nous pouvons vous aider.
Dans cet article de blog, nous expliquons les processus d’identification des protéines par LC-MS/MS. Nous décrirons comment préparer votre échantillon, comment fonctionne la machine, comment un programme peut identifier les protéines, et nous partagerons même notre protocole optimisé pour l’identification des protéines dans le gel. Vous serez en mesure d’identifier la protéine inconnue en un rien de temps.
Comment préparer votre échantillon pour l’identification des protéines par spectrométrie de masse ?
Cela dépend vraiment de la manière dont vous avez découvert que vous aviez une protéine à identifier. Avez-vous purifié votre protéine d’intérêt et trouvé une autre protéine éluée à partir de la préparation ? Ou avez-vous fait une immunoprécipitation de votre protéine cible et coloré le gel SDS PAGE pour trouver une bande principale en plus de votre appât ?
Dans tous les cas, vous devrez refaire le protocole avec du matériel propre pour limiter la contamination de votre échantillon par les kératines.
Ces protéines gênantes sont présentes partout et peuvent réellement entraver l’identification de votre protéine si elles sont trop concentrées dans votre échantillon.
Comme nous allons nous concentrer sur l’identification d’une protéine à partir d’un gel, nous vous suggérons de jeter un coup d’œil à notre protocole d’identification d’une protéine à partir d’un gel coloré au bleu de Coomassie pour avoir un exemple de l’expérimentation elle-même. Comme indiqué dans notre article sur la préparation des échantillons, vous devez toujours travailler avec des gants, des pointes de pipette filtrées et des tubes Eppendorf à faible liant. Cela améliorera grandement la récupération des peptides et la réussite de votre expérience. Quelle que soit la méthode de préparation des échantillons que vous utilisez, vous devez garder à l’esprit qu’en spectrométrie de masse, vous devez toujours travailler dans un environnement propre et avec des outils appropriés.
Comme vous le savez, les protéines sont en fait une chaîne d’acides aminés liés entre eux de manière covalente. Afin d’identifier votre protéine par LC-MS/MS, vous devez la décomposer en parties plus petites, appelées peptides. Cela se fait généralement par digestion enzymatique à l’aide d’enzymes protéolytiques, telles que la trypsine ou la chymotrypsine. Le choix de l’enzyme à utiliser est déterminé par la séquence de la protéine en question.
Dans le cas d’une protéine inconnue, nous choisissons généralement la trypsine en raison de son faible taux de mauvais clivage et de sa forte activité dans des tampons plutôt dénaturants. Là encore, le protocole de digestion enzymatique variera en fonction de la matrice dans laquelle se trouve votre protéine. Si elle se trouve dans un tampon à l’état relativement pur (provenant d’une purification par HPLC, par exemple), il est probable que vous puissiez simplement ajouter l’enzyme et procéder à la digestion. Si votre protéine se trouve dans un gel, la procédure peut être un peu plus complexe. Le résultat final devrait cependant toujours être le même : un mélange de peptides propre que vous pouvez analyser par LC-MS/MS.
Téléchargez votre protocole ici
Acquisition de données LC-MS/MS
Maintenant que vous avez votre mélange de peptides, vous êtes prêt à l’envoyer pour analyse à votre fournisseur de solutions en spectrométrie de masse préféré. Il existe plusieurs modes d’acquisition différents que vous pouvez utiliser pour identifier les protéines, mais nous nous concentrerons sur l’acquisition dépendante de l’information (IDA, également connue sous le nom d’acquisition dépendante des données, ou DDA) dans cet article. L’IDA utilise la vitesse et la résolution très élevées du spectromètre de masse pour déterminer la masse des peptides présents dans votre échantillon. Voici comment cela fonctionne :
2.1 – Séparation chromatographique (LC) :
Le mélange de peptides est chargé sur une colonne chromatographique. Les peptides dont la composition en acides aminés est différente auront des affinités différentes avec la colonne. En appliquant à la colonne des phases liquides dont la concentration en solvant organique augmente au fil du temps, nous pouvons séparer les peptides les uns des autres. En fonction de leur affinité pour la colonne, les peptides seront progressivement libérés de la colonne par l’augmentation de la concentration organique dans la phase mobile et entreront progressivement dans le spectromètre de masse. La séparation chromatographique permet de concentrer les peptides à un moment donné et facilite leur détection par l’appareil. En moyenne, sur une micro-LC avec un gradient de 60 minutes, il faut environ 45 secondes pour que toutes les copies d’un peptide soient éluées de la colonne.
2.2 – Analyse par spectrométrie de masse :
En spectrométrie de masse, le terme « cycle » représente la somme de toutes les petites tâches effectuées par la machine. Par exemple, en mode IDA, un cycle comprend un balayage de sondage et plusieurs balayages d’ions produits (jusqu’à 40 pour les spectromètres de masse à haute résolution). Chacun des composants d’un cycle prend un peu de temps pour être achevé. Par exemple, si un cycle comprend un balayage d’étude de 200 millisecondes et 40 balayages d’ions produits d’une durée de 35 millisecondes chacun, le temps total nécessaire à la machine pour effectuer un cycle est d’environ 1,6 seconde (200 ms + (40 x 35 ms) = 1600 ms). Une fois qu’un cycle est terminé, la machine commence un nouveau cycle. Ce processus se poursuit pendant toute la durée de l’analyse. Au cours d’un gradient de 60 minutes, le spectromètre de masse effectue environ 2118 cycles. Il n’est donc pas étonnant qu’une analyse par spectrométrie de masse produise autant de données ! Voici la description des deux principaux types de balayage nécessaires à une analyse en mode IDA.
Balayage d’enquête (une fois au début du cycle) : Pour identifier les protéines avec le mode IDA, le spectromètre de masse effectue d’abord une lecture complète du flux d’ions, appelée balayage d’enquête. Le balayage d’étude enregistre le rapport masse sur charge (m/z) de chaque ion qui entre dans la machine à ce moment-là. Le balayage dure environ 200 millisecondes. À la fin du balayage, l’appareil sait quels ions étaient présents dans l’échantillon à ce moment-là et peut les classer en fonction de l’intensité du signal.
Product ion scan (répété plusieurs fois) : la deuxième partie de la méthode IDA est connue sous le nom de balayage de l’ion produit. Au cours du balayage d’étude, la machine a créé une liste des x ions les plus intenses (jusqu’à 40) dans le flux d’ions entrant dans la machine. Le premier balayage de l’ion produit isole le premier ion le plus intense, le fragmente et enregistre le rapport m/z de tous ses fragments. Cette opération dure environ 35 millisecondes. Une fois cette opération terminée, l’appareil exécute un deuxième balayage d’ions de produit. Elle isole, fragmente et enregistre le rapport m/z des fragments du deuxième ion le plus intense… et ainsi de suite jusqu’à ce que le nombre d’ions à analyser dans le cycle soit atteint (jusqu’à 40 dans un cycle). Ensuite, le premier cycle se termine et le deuxième cycle commence. Un peptide élue de la colonne pendant environ 45 secondes dans un gradient LC de 60 minutes. Sachant qu’un cycle ne dure que 1,6 seconde, cela signifie que nous pourrions potentiellement détecter le même peptide environ 28 fois pendant son élution de la colonne (45 sec/1,6 sec par cycle = 28). Étant donné que la machine n’enregistre que les 40 ions les plus intenses sur plusieurs centaines à la fois, nous pourrions potentiellement manquer beaucoup d’informations. C’est ici qu’intervient une fonction d’exclusion utile de la méthode IDA. En effet, nous pouvons demander à l’appareil de n’enregistrer le même ion que deux fois et de l’exclure pendant un certain nombre de secondes par la suite. Au cours du cycle suivant, lorsque cet ion est détecté dans le balayage d’étude après avoir été enregistré deux fois, il ne sera pas compté dans les 40 balayages d’ions produits. Au lieu de cela, la machine isolera et fragmentera le 40+1e ion le plus intense. Ce processus peut se poursuivre indéfiniment, ce qui explique comment la machine peut obtenir une bonne profondeur de séquençage, même si elle est limitée à 40 balayages d’ions produits par cycle !
Identification des protéines
Le résultat final de la méthode IDA est une liste d’ions précurseurs (enregistrés pendant le balayage de l’enquête) et de leurs fragments associés (enregistrés pendant le balayage de l’ion produit). Les fichiers produits par la spectrométrie de masse sont ensuite introduits dans un programme qui effectuera l’identification des protéines en deux étapes*.
*Nous travaillons avec le logiciel ProteinPilot (de Sciex) et nous verrons comment cette suite d’algorithmes fonctionne pour l’identification de protéines. Notez qu’il existe d’autres outils qui peuvent effectuer des analyses similaires, tous avec de légères différences dans les résultats.
3.1 – Identification des peptides :
Dans ProteinPilot, l’algorithme chargé de l’identification des peptides s’appelle Paragon. Pour identifier les peptides, ce programme examine tous les m/z des ions produits afin de trouver des fragments peptidiques très courts pouvant correspondre à 2 ou 3 acides aminés. Ces petits fragments, appelés taglets, peuvent facilement être identifiés avec une bonne confiance car le nombre de modifications post-traductionnelles différentes pouvant être trouvées sur un peptide aussi petit est limité. Pour déterminer leur séquence, le programme fait correspondre le m/z détecté du taglet au m/z théorique de tous les acides aminés, avec et sans modifications. Si le m/z du taglet se situe dans une petite plage de la séquence théorique d’un petit peptide, il est considéré comme identifié et reçoit un bon score. En revanche, si la différence entre le m/z du taglet et une séquence théorique est plus importante, il recevra un score plus faible. Ce processus peut être visualisé dans la partie supérieure de la figure 2 ci-dessous.
Une fois que la séquence d’acides aminés du taglet est déterminée, Paragon l’associe à toutes les protéines de la base de données afin de déterminer où cette petite séquence peut être trouvée. Une fois que toutes les étiquettes ont été cartographiées, le programme attribue une température à chaque région du protéome en fonction de la densité d’étiquettes qu’elle contient.
Par exemple, si une région du protéome contient la séquence de plusieurs marqueurs ou de marqueurs ayant des scores très élevés, elle sera identifiée comme chaude par le programme et bénéficiera d’une plus grande puissance de recherche (expliqué plus en détail dans la figure 2). À l’inverse, une région qui ne contient qu’un seul ou aucun taglet sera identifiée comme froide. La logique sous-jacente est que si plusieurs marqueurs se rapportent à une région spécifique, cela signifie que les peptides qui contiennent le marqueur se rapportent également à cette région, ce qui la rend plus digne d’une puissance de recherche élevée. L’utilisation de cette stratégie permet à l’algorithme de rechercher plus efficacement, puisqu’il consacrera moins d’efforts aux régions froides du protéome, c’est-à-dire là où l’on s’attend à trouver moins de peptides, qu’aux régions chaudes. Les recherches effectuées avec l’algorithme Paragon sont donc plus rapides qu’avec d’autres algorithmes, tout en conservant la même efficacité. En résumé, la carte de température du protéome qui a été générée à partir de la cartographie du taglet au protéome est utilisée par Paragon pour déterminer la puissance de recherche qui sera attribuée pour compléter la séquence de chaque taglet et trouver le peptide dont il est issu. Une très belle vidéo expliquant l’algorithme Paragon est disponible ici.

Fig2: Example de recherche de Paragon.
Les différentes étiquettes identifiées par Paragon sont énumérées en haut de la figure. Plus la ligne rouge sous le taglet est épaisse, plus le score obtenu est élevé. Dans cet exemple, une seule protéine est représentée. Les lignes rouges sous la séquence indiquent la correspondance entre les marqueurs et la séquence d’acides aminés de la protéine. Plus une région possède de taglets, plus elle est considérée comme « chaude » par Paragon. Une fois ce processus terminé, Paragon attribue une puissance de recherche différente aux régions, en fonction de leur « température » relative. Si une région est « chaude », Paragon effectuera des recherches plus approfondies pour trouver des peptides dans cette région. Par exemple, une région « chaude » fera l’objet d’une recherche pour toutes les modifications post-traductionnelles connues à ce jour et pour un grand nombre de mauvais clivages rares, tandis qu’une région « froide » ne fera l’objet d’une recherche que pour les PTM les plus courantes. Cette figure est tirée de l’article original de Paragon publié dans Sciex (2007).
3.2- Identification d’une protéine:
L’algorithme d’identification des protéines de SCIEX est appelé Pro Group™ Algorithm. Le but de ce programme est de prendre les peptides identifiés par Paragon et d’en déduire la liste la plus probable de protéines expliquées par cette évidence peptidique. Bien que la mise en correspondance d’une séquence peptidique avec une protéine puisse sembler plus simple que la détermination de la séquence des peptides à partir de ses fragments, l’identification des protéines présente des difficultés fondamentales que le programme doit surmonter. Tout d’abord, de nombreuses protéines partagent des régions conservées dans leur séquence d’acides aminés. Par conséquent, de nombreux peptides peuvent provenir de plus d’une protéine. En outre, certains spectres MS/MS ne peuvent être attribués à une seule séquence. Il y a donc beaucoup d’incertitudes à prendre en compte lors de la détermination de la liste des protéines d’un échantillon. Prenons un exemple où un peptide X peut être trouvé dans 3 protéines différentes, Prot1, Prot2 et Prot3. Lors de la détection de ce peptide, le programme n’a aucun moyen de savoir, d’un point de vue biologique, si le peptide provient de Prot1, Prot2, Prot3 ou d’une combinaison de ces protéines. Par conséquent, tous les peptides partagés, comme le peptide X, seront associés à une seule de ces protéines, Pept1 par exemple. Ainsi, dans la liste de protéines qui sera générée par le programme, nous ne trouverons que Prot1 (à moins qu’un peptide de différenciation entre Prot1 et Prot2 ou Prot3 puisse être détecté, dans ce cas, les deux protéines seront affichées).
À quoi ressemble l’identification d’une protéine ?
En suivant le processus décrit dans cet article, la bande inconnue sur votre gel est maintenant une protéine identifiée. Il est intéressant de noter que le même processus de spectrométrie de masse peut être couplé à d’autres techniques de préparation d’échantillons, telles que l’immunoprécipitation, pour identifier les partenaires d’interaction protéine-protéine ou les modifications post-traductionnelles de votre protéine d’intérêt.
Les captures d’écran ci-dessous illustrent l’identification d’une protéine dans le logiciel ProteinPilot. Elles soulignent également la complexité d’une telle analyse et la quantité de données qui peuvent être générées avec une seule expérience de spectrométrie de masse !
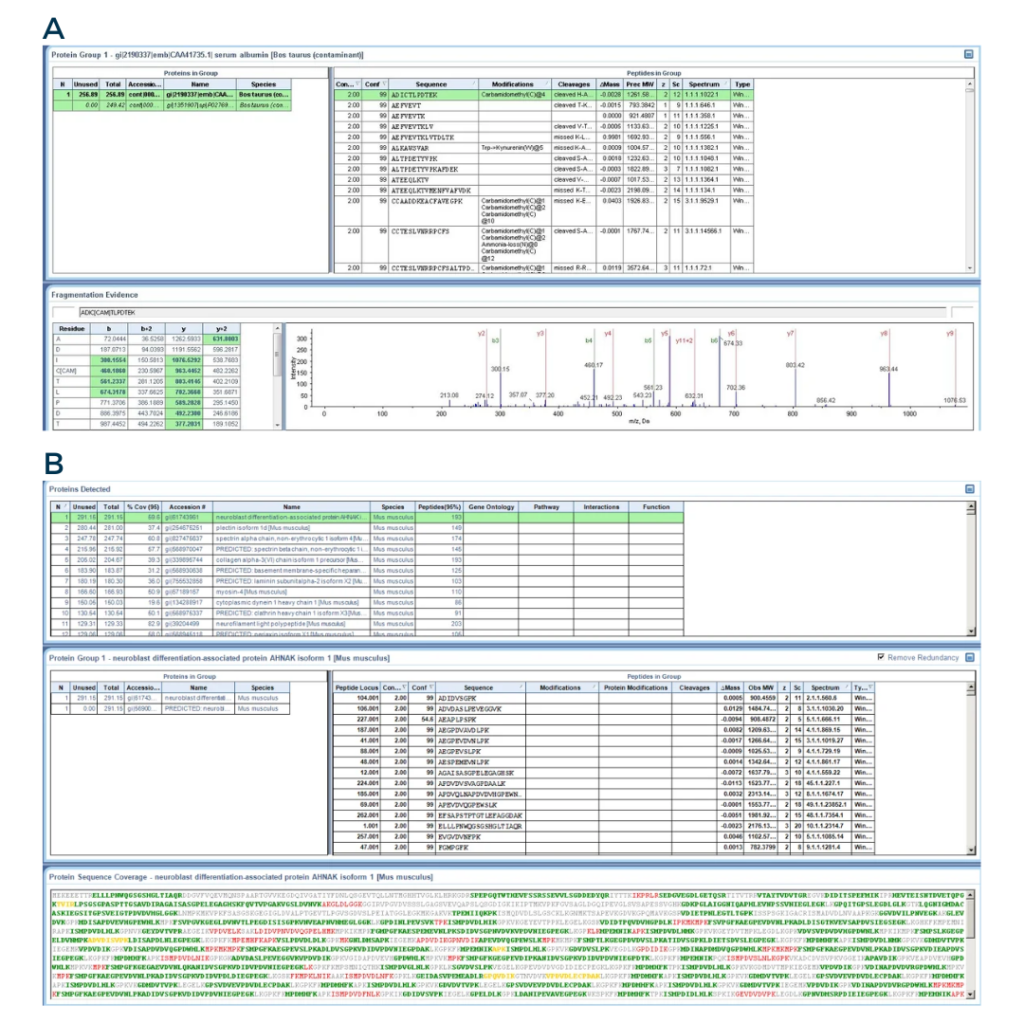
Fig3 : à partir du logiciel ProteinPilot
A) Capture d’écran d’une identification de peptide. Le panneau inférieur droit affiche les spectres MS/MS d’un peptide.
B) Capture d’écran d’une identification de protéine. Le panneau inférieur montre les peptides qui ont été détectés lors de l’identification des peptides. Les lettres vertes représentent les peptides dont la confiance est bonne, tandis que les lettres jaunes et rouges représentent les peptides dont la confiance est moyenne et faible, respectivement.
Conclusion
La spectrométrie de masse est un outil puissant pour identifier les protéines d’un échantillon. Qu’il s’agisse d’un échantillon très complexe ou d’une seule bande de protéine découpée dans un gel de polyacrylamide, la LC-MS/MS est le moyen le plus efficace de faire avancer votre projet de protéomique.
D’autres questions sur cet article de blog ou sur la façon dont la spectroscopie de masse peut vous aider dans votre recherche ?